
Il nuovo Regolamento dei Dispositivi Medici. Cosa cambia? di Luca Roberti
I Dispositivi Medici sono importanti strumenti per la nostra vita quotidiana e per la nostra salute, pensiamo al termometro per la temperatura corporea, le lenti oftalmiche e a contatto, le montature per lenti correttive, i cerotti, materassi ortopedici, pannoloni per incontinenza, i ventilatori, l’aereosol o i dispositivi impiantabili, tra i dispositivi medici in vitro troviamo i contenitori dei campioni, le strisce, i vari test e autotest.
La data del 21 maggio 2021 ha segnato una tappa importante per questo processo normativo Europeo,
in quanto ha visto l’entrata in vigore del nuovo regolamento dei Dispositivi Medici (MDR) il 745/2017/EU (1) e del 746/2017/EU (dispositivi medico diagnostici in vitro IVDR). Questo nuovo regolamento riduce i rischi di possibili discrepanze interpretative nella Comunità Europea. La classificazione dei DM viene divisa in 4 classi (vedi foto): Classe I, Classe IIa, Classe IIb e Classe III e per IDVR da Classe A, B, C e D.
Sono state rafforzate le norme sulle indagini cliniche per i dispositivi medici e gli studi delle prestazioni per i dispositivi medici in vitro. Le nuove regole descrivono chiaramente come queste indagini devono essere progettate, notificate e/o autorizzate, condotte, registrate e comunicate.
Per i produttori, i regolamenti aggiungono nuovi requisiti e rafforzano quelli esistenti. I produttori devono mettere in atto sistemi per la gestione del rischio e della qualità, condurre valutazioni cliniche o delle prestazioni, redigere documentazione tecnica e 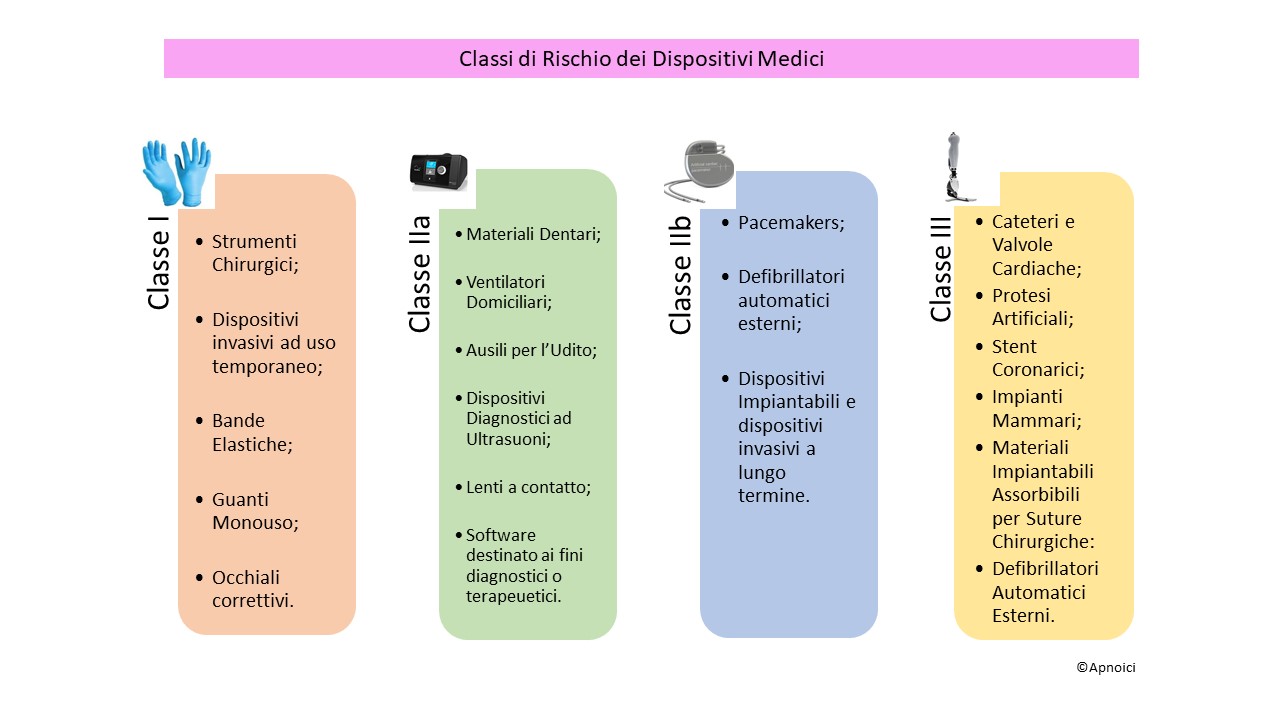 mantenere tutto aggiornato.
mantenere tutto aggiornato.
I fabbricanti sono tenuti ad applicare procedure di valutazione della conformità per immettere i propri dispositivi sul mercato. Il livello di evidenza clinica necessaria per dimostrare la conformità di un dispositivo dipende dalla sua classe di rischio.
Il regolamento chiarisce anche la distinzione tra vigilanza e sorveglianza post-commercializzazione.
Il primo include l’identificazione e la segnalazione di incidenti gravi e l’esecuzione di azioni correttive relative alla sicurezza. Richiede una cooperazione diretta ed efficiente tra operatori sanitari, istituzioni sanitarie, produttori e autorità nazionali competenti per i dispositivi medici. La sorveglianza post-commercializzazione comporta il monitoraggio delle informazioni disponibili per riconfermare periodicamente che i vantaggi del dispositivo continuano a prevalere sui rischi. I regolamenti aggiungono regole più rigorose per la designazione degli Organismi Notificati con valutazioni indipendenti dai fabbricanti e dai loro dispositivi (MDR/IVDR).
L’Organismo Notificato “ON” che certifica il DM, tra i vari compiti, si occupa di valutare il sistema di gestione della qualità del produttore; valutare la documentazione tecnica talvolta unitamente alla verifica del campione di prodotto; rilasciare certificati di marcatura CE; predisporre audit di sorveglianza annuali, audit senza preavviso almeno ogni 5 anni, con prove a campione; revisionare la sorveglianza post-commercializzazione.
Durante il periodo transitorio, che terminerà il 26 maggio 2024, gli Organismi Notificati “ON” dovranno applicare le nuove regole e ri-certificare tutti i Dispositivi in commercio. Attualmente in Europa sono 33 gli ON, e i DM da certificare sono circa 20.000.. Questi grossi volumi stanno preoccupando i costruttori e i distributori e viene richiesto dagli stessi costruttori un ulteriore periodo di slittamento delle date di scadenza delle vecchie certificazioni per evitare un eventuale blocco nel 2024.
Il primo caso importante di Recall (richiamo) mondiale (negli USA) e di Avviso di Sicurezza (in Europa) è molto recente (giugno 2021) e riguarda la schiuma fonoassorbente contenuta in più di 15 milioni di ventilatori prodotti dalla Philips-Respironics tra il 2010 e il 2021.
Questo richiamo spontaneo ha fatto emergere prepotentemente le numerose falle nel sistema di controllo, per la mancanza di un rodato sistema di dispositivo di vigilanza. Negli ultimi mesi sono emerse anche importanti criticità sulla carenza di componentistica in particolar modo nei medical device, dovuto all’aumento dei costi per la produzione, logistica e riguarda la carenza dei Semi-conduttori e dei materiali plastici.
Si è evidenziata l’urgente necessità di creare in altri continenti oltre che in Asia, nuovi siti produttivi. Gli Stati Uniti hanno risposto a questa crisi di approvvigionamento ed approvato in questi giorni una specifica Legge il “Chips and Science Act” che prevede la costruzione di nuove fabbriche e infrastrutture per rendere gli USA indipendenti dai produttori asiatici.
Un nuovo Decreto del 31 marzo 2022 (2),
voluto dal Sottosegretario Sileri, istituisce la “Rete Nazionale della dispositivo-vigilanza” con il relativo sistema informativo, la rete è stata istituita per favorire lo scambio tempestivo delle informazioni tra il Ministero e le Regioni, riguardanti gli incidenti e le azioni di sicurezza che coinvolgono i dispositivi medici e i dispositivi medico diagnostici in vitro.
Per approfondimenti:
1 – https://health.ec.europa.eu/medical-devices-new-regulations/overview_en
2 – https://www.salute.gov.it/portale/temi/p2_6.jsp?lingua=italiano&id=90&area=dispositivi-medici&menu=vigilanza
https://youtu.be/YhngxYmW0jU (webinar con ON ed esperti in Dispositivi Medici)
https://www.confindustriadm.it/il-settore-in-numeri-2020/
Autore dell’articolo*: Luca Roberti – Presidente Associazione Apnoici Italiani e Paziente Esperto Eupati

*Seguirà una seconda parte in cui verranno approfonditi i temi del coinvolgimento dei Pazienti nella Gare e della nascita dell’Agenzia di MD.




